Glutathione: Discovery and Research History
How glutathione went from an unnamed reducing substance in 1888 yeast extracts to a fully mapped tripeptide with three enzyme families and a redox-signaling role, told through the investigators who defined it. Educational reference.

For research use only. Not for human consumption. This article is educational reference material. It is not medical advice and is not a recommendation to use any substance.
Glutathione: A Timeline of Its Discovery and Scientific Development
Few small molecules have a documentation trail as long or as contested as glutathione (GSH; γ-L-glutamyl-L-cysteinyl-glycine). Its history is unusual among the compounds in the Sparta Labs library because it was described, mis-described, renamed, structurally revised, synthesized, and then re-interpreted across roughly 130 years of literature. This article traces that development not as a generic timeline but through the specific puzzles that occupied each generation of investigators: what the reducing substance in yeast actually was, why its peptide bond refused to behave like a normal one, and how a single molecule ended up at the center of three separate enzyme families. The chemistry and verification standards for research-grade glutathione are treated separately in the glutathione sourcing and quality reference.
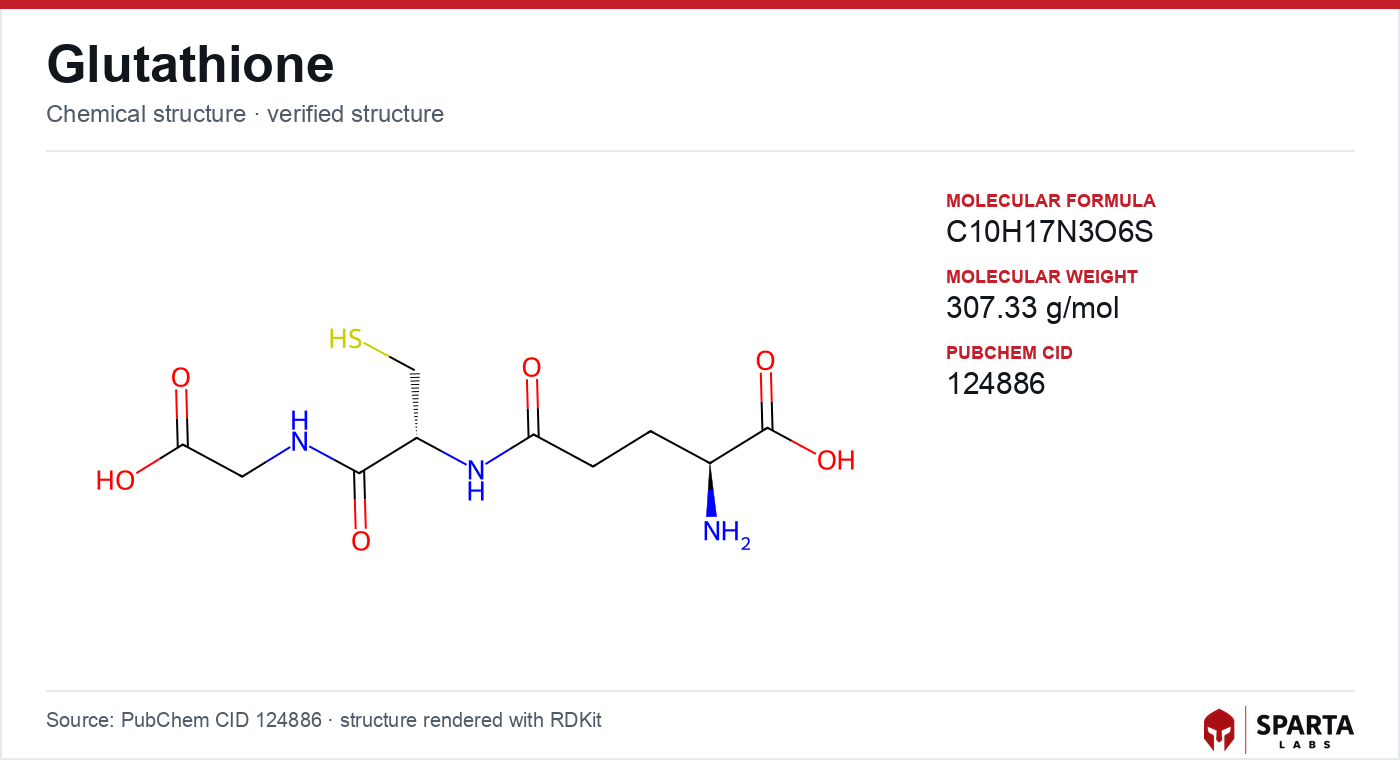
Figure: chemical structure of Glutathione (γ-L-glutamyl-L-cysteinyl-glycine).
An unnamed reducing substance: de Rey-Pailhade, 1888
The earliest entry in the glutathione record does not use the modern name at all. In 1888 the French chemist J. de Rey-Pailhade, working in Toulouse, isolated a water-soluble substance from brewer's yeast, muscle, and other tissues that had one striking property: it reduced elemental sulfur to hydrogen sulfide. Lacking any structural handle on the molecule, he named it for its behavior — "philothion," from the Greek for "love of sulfur" [1].
De Rey-Pailhade's observation is worth pausing on, because it correctly identified thiol chemistry three decades before anyone could characterize the responsible group. What he had detected was the free sulfhydryl (–SH) of the cysteine residue, the reactive center that would eventually explain nearly everything glutathione does. The compound then effectively disappeared from the literature for a generation, an early example of an experimental result outrunning the analytical tools needed to interpret it.
The Hopkins isolation and the dipeptide error: 1921–1929
The canonical rediscovery belongs to Frederick Gowland Hopkins at Cambridge. In 1921 Hopkins isolated a thiol-containing compound from yeast and animal muscle, noted that it cycled readily between reduced and oxidized forms, and coined the name "glutathione" to reflect its glutamic acid and sulfur (thiol) content [1]. His initial characterization, however, contained a now-famous error: Hopkins reported glutathione as a dipeptide of glutamate and cysteine, with glycine absent from his structure.
The correction is one of the more instructive episodes in early biochemistry. Independent work through the mid-1920s, including analyses that found glycine in purified preparations, pressed Hopkins to re-examine his material with improved purification. In 1929 he published a revised structure establishing glutathione as a tripeptide — glutamate, cysteine, and glycine — resolving the discrepancy [1]. That same year Hopkins shared the Nobel Prize in Physiology or Medicine, awarded for his broader work on accessory food factors (vitamins), with the glutathione research standing as a prominent thread in his legacy. Findings from research models do not establish safety or efficacy in humans. Sparta Labs makes no claims about the use of this compound.
The gamma-linkage puzzle: Harington and Mead, 1935
Knowing the three amino acids was not the same as knowing how they were joined. The decisive structural work came from Charles Robert Harington and Thomas Hobson Mead, who reported a total chemical synthesis of glutathione in 1935 and in doing so confirmed the connectivity as γ-L-glutamyl-L-cysteinyl-glycine [1].
The critical detail their synthesis nailed down was the γ-peptide bond: glutamate is attached to cysteine not through its α-carboxyl group, as in ordinary proteins, but through the carboxyl on its side chain (the γ-carboxyl). This is a genuinely unusual arrangement. Standard cellular proteases recognize α-peptide bonds; the γ-linkage is invisible to most of them, which is a large part of why glutathione persists at high concentration inside cells rather than being rapidly digested. The cysteine–glycine bond, by contrast, is a conventional α-linkage. That asymmetry — one non-standard bond and one standard bond — turns out to govern both the molecule's stability and the way specialized enzymes assemble and dismantle it. This structural specificity is a recurring theme in the glutathione mechanism of action literature.
Three enzymes, three research threads: 1937–1970s
What distinguishes glutathione's history from that of most peptides is that its functional characterization did not converge on one pathway. Instead, three largely separate enzyme lineages were worked out over several decades, each anchoring its own field.
Thread one — the glutathione S-transferases
Beginning with the recognition that animals excrete "mercapturic acids," investigators including Booth, Boyland, and Sims described in the 1950s the enzymatic conjugation of glutathione to electrophilic and aromatic compounds [2]. This established the glutathione S-transferase (GST) family and placed GSH at the heart of xenobiotic and drug metabolism, a role that later made GST expression a subject of pharmacology and toxicology research.
Thread two — glutathione peroxidase and the selenium surprise
In 1957 Gordon C. Mills reported an erythrocyte enzyme that reduced hydrogen peroxide using glutathione as the electron donor, naming it glutathione peroxidase (GPx) [3]. A second surprise followed years later: GPx was found to be a selenoenzyme, incorporating selenium as selenocysteine in its active site. That discovery connected a trace element to glutathione biochemistry and seeded decades of work on selenium-dependent antioxidant enzymes and their multiple tissue-specific isoforms.
Thread three — the γ-glutamyl cycle and Alton Meister
The most concentrated single-laboratory contribution came from Alton Meister at Cornell University Medical College. In the early 1970s Meister and colleagues proposed the γ-glutamyl cycle, a framework in which glutathione participates in membrane-associated amino acid transport through the sequential action of γ-glutamyl transpeptidase (GGT) at the cell surface and resynthesis inside the cell [4]. Meister's group went on to characterize the biosynthetic enzymes — glutamate-cysteine ligase (GCL) and glutathione synthetase — and to develop specific inhibitors that became standard experimental tools. The comprehensive Meister and Anderson review of 1983 remains a foundational reference for the entire field [4].
From genes to redox signaling: 1980s–present
Molecular cloning reoriented glutathione research in the 1980s and 1990s. The catalytic (GCLC) and modifier (GCLM) subunits of glutamate-cysteine ligase were cloned, enabling study of the enzyme's transcriptional control and the construction of genetically modified animal models. GCLM-knockout mice proved viable but showed markedly lower tissue GSH and heightened sensitivity to oxidative insults in experimental settings, which helped define GSH as an essential endogenous biomolecule.
A regulatory layer emerged with the identification of the Nrf2–Keap1 system. Work from Masayuki Yamamoto's laboratory and others showed that the transcription factor Nrf2 binds antioxidant response elements (AREs) in the promoters of GCLC, GCLM, and many other genes, providing a transcriptional basis for adaptive changes in GSH biosynthesis under oxidative conditions [5]. This placed glutathione within a much larger stress-response network rather than treating it as an isolated antioxidant.
Two more recent developments have kept the molecule at the research frontier. First, S-glutathionylation was characterized as a reversible post-translational modification, with proteomic surveys identifying hundreds of candidate protein targets and the glutaredoxin enzymes reported to add and remove the modification — recasting glutathione as a redox-signaling participant, not merely a scavenger [2]. Second, the enzyme GPx4 was identified as a central regulator of ferroptosis, a form of regulated cell death driven by lipid peroxide accumulation; because GPx4 uses GSH to reduce membrane lipid hydroperoxides, the glutathione system was drawn directly into one of the most active areas of contemporary cell-death and cancer biology [3].
The theme of age-associated decline in an endogenous cofactor connects glutathione to a broader research cluster; a parallel trajectory has developed around NAD+, another intracellular metabolite studied for changes across the lifespan, and the glutathione research overview situates GSH within that comparative context.
Why the history matters for interpretation
Reading the glutathione literature productively requires keeping its layered history in mind. A single molecule accumulated meanings sequentially — reducing agent (1888), tripeptide (1929), γ-linked structure (1935), detoxification cofactor, peroxide substrate, transport intermediate, transcriptionally regulated metabolite, and signaling modification — and papers from different eras use "glutathione" against different conceptual backdrops. For researchers evaluating the compound as a laboratory reference material, the current glutathione product page and the accompanying sourcing and quality reference describe the analytical standards used to verify identity and purity of the tripeptide those investigators spent a century defining.
References
-
Meister A. On the discovery of glutathione. Trends Biochem Sci. 1988;13(5):185-188. PMID: 3076280. DOI: 10.1016/0968-0004(88)90148-X. Link
-
Aquilano K, Baldelli S, Ciriolo MR. Glutathione: new roles in redox signaling for an old antioxidant. Front Pharmacol. 2014;5:196. PMID: 25206336. DOI: 10.3389/fphar.2014.00196. Link
-
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175-185. PMID: 31981735. DOI: 10.1016/j.freeradbiomed.2020.01.027. Link
-
Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711-760. PMID: 6137189. DOI: 10.1146/annurev.bi.52.070183.003431. Link
-
Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30(1-2):1-12. PMID: 18796312. DOI: 10.1016/j.mam.2008.08.006. Link
Disclaimer. Statements in this article have not been evaluated by the Food and Drug Administration. This compound is not intended to diagnose, treat, cure, or prevent any disease. Sparta Labs sells research-use-only materials. Content is provided for educational and informational purposes only and does not constitute medical advice. Consult a qualified medical professional for any health concerns.
Frequently asked questions
What was glutathione originally called when it was first described?
In 1888 the French chemist J. de Rey-Pailhade named the substance "philothion," Greek for "love of sulfur," after observing that it reduced elemental sulfur to hydrogen sulfide in yeast and tissue extracts. He detected the reactive thiol group decades before its chemistry could be characterized. The modern name "glutathione" was introduced later by Frederick Gowland Hopkins.
Why did Hopkins first describe glutathione as a dipeptide?
Hopkins' 1921 preparations led him to report glutathione as a dipeptide of glutamate and cysteine, with glycine absent. Improved purification and independent analyses through the mid-1920s identified glycine in the molecule, and Hopkins published a corrected tripeptide structure in 1929.
What is unusual about glutathione's peptide bonds?
Glutamate is joined to cysteine through a gamma-peptide bond, using glutamate's side-chain carboxyl rather than the standard alpha-carboxyl found in ordinary proteins. Harington and Mead confirmed this in their 1935 total synthesis. The gamma-linkage is not recognized by most cellular proteases, which is a major reason glutathione persists at high intracellular concentration.
How did one molecule end up central to three different enzyme families?
Its functional characterization proceeded along three separate threads: the glutathione S-transferases described in xenobiotic metabolism, glutathione peroxidase reported by Gordon Mills in 1957 and later found to be a selenoenzyme, and the gamma-glutamyl cycle framework proposed by Alton Meister's laboratory in the early 1970s. Each thread anchored its own research field.
Why is glutathione still an active area of modern research?
Twenty-first-century work characterized S-glutathionylation as a reversible redox-signaling modification and identified the enzyme GPx4, which uses glutathione, as a central regulator of ferroptosis. These findings connected the century-old molecule to contemporary cell-death and cancer biology, keeping it at the research frontier.