Semaglutide: A Research Overview
A structure-first reference on semaglutide, tracing how fatty-diacid acylation, a DPP-4-resistant Aib substitution, and the SNAC oral-delivery system emerged from three decades of GLP-1 analogue engineering at Novo Nordisk.

For research use only. Not for human consumption. This article is educational reference material. It is not medical advice and is not a recommendation to use any substance.
Introduction
Semaglutide is a synthetic glucagon-like peptide-1 (GLP-1) receptor agonist: a 31-amino-acid peptide engineered to reproduce the receptor-binding activity of the endogenous incretin hormone GLP-1(7-37) while resisting the rapid clearance that limits the native molecule. It was developed by a research team at Novo Nordisk and first described in detail in the peer-reviewed literature in 2015 [1]. What distinguishes semaglutide as a chemistry problem is not its receptor target, which it shares with several earlier analogues, but the set of molecular design choices that pushed its plasma half-life from minutes to days. This overview is organized around those design choices rather than a generic template, tracing the specific structural engineering that defines the compound.
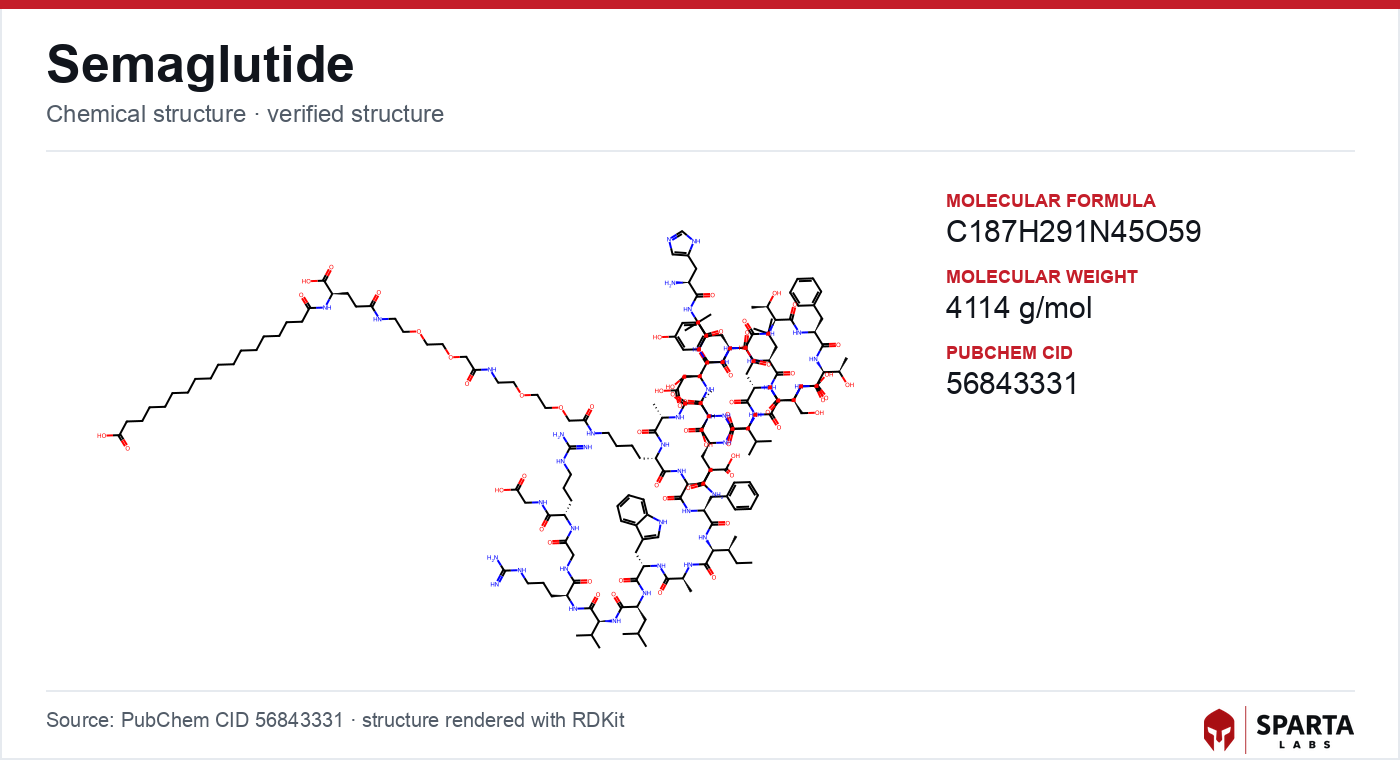
Figure: chemical structure of Semaglutide.
The half-life problem that shaped the molecule
Every structural feature of semaglutide traces back to a single pharmacokinetic constraint. Glucagon-like peptide-1 was identified as a potent insulinotropic factor in the 1980s. Mojsov, Weir, and Habener reported in 1987 that GLP-1(7-37), a cleavage product of proglucagon expressed in intestinal L-cells, stimulated insulin secretion from the perfused rat pancreas at picomolar concentrations [2]. Holst and colleagues independently characterized the truncated peptide as an insulin-releasing hormone from the distal gut in the same year [3].
Findings from research models do not establish safety or efficacy in humans. Sparta Labs makes no claims about the use of this compound.
The molecule that emerged from this foundational work carried a disabling flaw as a therapeutic scaffold: native GLP-1 is degraded almost immediately by the serine protease dipeptidyl peptidase-4 (DPP-4), which cleaves the peptide between residues 8 and 9 and confers a circulating half-life of under two minutes [4]. Any durable GLP-1 analogue therefore had to solve two problems at once: block the DPP-4 cleavage site, and slow renal filtration of a small peptide that the kidney would otherwise clear rapidly. Semaglutide's structure is best read as two engineered answers to those two problems.
Structure and the engineering of persistence
The primary structure and synthesis of semaglutide were described by Lau, Bloch, Schäffer, Knudsen, and colleagues in a 2015 paper in the Journal of Medicinal Chemistry [1]. The compound is a 31-amino-acid analogue of human GLP-1(7-37) with three deliberate modifications, each addressing a defined liability of the native sequence.
The first modification protects the N-terminus. The alanine at position 8, the residue immediately adjacent to the DPP-4 cleavage bond, is replaced by alpha-aminoisobutyric acid (Aib), a non-canonical, doubly methylated amino acid whose steric bulk blocks protease access [1]. This single substitution addresses the proteolytic half of the half-life problem.
The second and third modifications address clearance and stability. The lysine at position 34 is substituted with arginine to remove an alternative acylation site, ensuring that the lipid anchor attaches only where intended [1]. The lysine at position 26 is then derivatized through a bifunctional linker composed of two gamma-glutamic acid spacers and a short polyethylene-glycol (mini-PEG) segment, terminating in a C18 fatty diacid moiety [1]. This acyl side chain binds reversibly and tightly to serum albumin, effectively cloaking the peptide within a large carrier protein that the kidney does not filter. Lau and colleagues reported that this design extends the terminal half-life in humans to approximately 165 hours [1]. The reported molecular formula is C₁₈₇H₂₉₁N₄₅O₅₉, with a molecular weight of approximately 4,114 daltons [1].
The GLP-1 receptor that semaglutide targets belongs to the class B subfamily of G-protein-coupled receptors, characterized by a large extracellular N-terminal domain that participates in ligand recognition [4]. The downstream signaling consequences of this binding are treated separately in the companion semaglutide mechanism-of-action article.
Acylation as a design lineage
Semaglutide did not appear in isolation; it is the second-generation product of a specific engineering strategy. The albumin-binding fatty-acid approach was introduced by the earlier once-daily analogue liraglutide, which used a shorter C16 fatty-acid chain attached through a single glutamate spacer [5]. Knudsen and Lau, in a 2019 review of both compounds, describe semaglutide as a redesign of that anchor: lengthening the lipid to a C18 diacid and elaborating the linker increased albumin affinity and slowed clearance further, converting a once-daily molecule into a once-weekly one [5].
This lineage places semaglutide within a broader family of engineered incretin analogues that researchers often study comparatively. A structurally distinct approach appears in the dual GIP/GLP-1 receptor agonist covered in the Tirzepatide research overview, while the amylin analogue described in the Cagrilintide research overview illustrates a different acylation target within the same metabolic-peptide space. Comparing the anchor chemistry across these molecules is one of the clearer ways to see how small lipid and linker changes translate into large pharmacokinetic differences.
The oral formulation and the SNAC system
A second, less obvious piece of semaglutide chemistry concerns delivery rather than the peptide itself. Peptides are generally destroyed in the gastrointestinal tract and absorbed poorly, which historically restricted GLP-1 analogues to injectable formats. The oral tablet formulation of semaglutide co-formulates the peptide with sodium N-[8-(2-hydroxybenzoyl)amino]caprylate (SNAC), an absorption enhancer [6]. Published research describes SNAC as buffering local gastric pH to reduce peptic degradation and transiently increasing gastric-epithelial permeability in the immediate vicinity of the tablet, creating a localized window for absorption [6]. Husain and colleagues reported cardiovascular-outcome data for the oral formulation in the PIONEER 6 trial [6]. This delivery chemistry, rather than any change to the peptide sequence, is what made an orally administered GLP-1 receptor agonist feasible.
Regulatory profile as a research landmark
Semaglutide is one of the more fully characterized compounds in its class, with a regulatory record spanning multiple formulations. A subcutaneous formulation received FDA approval in December 2017 as an adjunct to diet and exercise in adults with type 2 diabetes mellitus. An oral tablet formulation followed in September 2019 for the same glycemic-management indication, the first oral GLP-1 receptor agonist to receive FDA authorization for any use. A higher-concentration subcutaneous formulation received FDA approval in June 2021 for chronic weight management in adults with obesity or overweight with a weight-related comorbidity. A cardiovascular risk-reduction indication was subsequently added following the SELECT trial, reported by Lincoff and colleagues in 2023 [7].
The compounding-relevant chronology is also part of the public record: in February 2025 the FDA issued a declaratory order resolving the previously declared shortage of semaglutide injection products, with enforcement-discretion periods for 503A and 503B compounding concluding later that spring [8]. The full sequence of regulatory milestones is covered in the semaglutide discovery and regulatory history article, and quality-control considerations for research material are treated in the semaglutide sourcing and verification reference. Research-grade semaglutide from Sparta Labs is offered with third-party-verified purity documentation for qualified laboratory applications.
Scientific lineage
The intellectual foundation of semaglutide predates the molecule by roughly three decades. Joel Habener and Svetlana Mojsov at Massachusetts General Hospital, and Jens Holst and colleagues in Denmark, independently established the insulinotropic properties of GLP-1 cleavage products in the 1980s [2, 3]. Daniel Drucker's laboratory later characterized the molecular control of proglucagon expression and the broader physiology of GLP-1 signaling [4]. Applied work at Novo Nordisk through the 1990s and 2000s, including contributions from Lotte Bjerre Knudsen, translated this biology into the acylation strategy that produced first liraglutide and then semaglutide [5]. The structure-activity reasoning behind semaglutide was set out formally by Lau et al. in 2015 [1]. In 2024, Habener, Mojsov, and Knudsen received the Albert Lasker Award for Basic Medical Research for their foundational contributions to the GLP-1 field [9].
References
-
Lau J, Bloch P, Schäffer L, et al. Discovery of the Once-Weekly Glucagon-Like Peptide-1 (GLP-1) Analogue Semaglutide. J Med Chem. 2015;58(18):7370–7380. doi:10.1021/acs.jmedchem.5b00726. PubMed PMID: 26308095. Link{target="_blank" rel="noopener noreferrer"}
-
Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. 1987;79(2):616–619. doi:10.1172/JCI112855. PubMed PMID: 3543057. Link{target="_blank" rel="noopener noreferrer"}
-
Holst JJ, Orskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 1987;211(2):169–174. doi:10.1016/0014-5793(87)81430-8. PubMed PMID: 3542566. Link{target="_blank" rel="noopener noreferrer"}
-
Drucker DJ. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018;27(4):740–756. doi:10.1016/j.cmet.2018.03.001. PubMed PMID: 29617641. Link{target="_blank" rel="noopener noreferrer"}
-
Knudsen LB, Lau J. The Discovery and Development of Liraglutide and Semaglutide. Front Endocrinol (Lausanne). 2019;10:155. doi:10.3389/fendo.2019.00155. PubMed PMID: 31031702. Link{target="_blank" rel="noopener noreferrer"}
-
Husain M, Birkenfeld AL, Donsmark M, et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N Engl J Med. 2019;381(9):841–851. doi:10.1056/NEJMoa1901118. PubMed PMID: 31185157. Link{target="_blank" rel="noopener noreferrer"}
-
Lincoff AM, Brown-Frandsen K, Colhoun HM, et al. Semaglutide and Cardiovascular Outcomes in Obesity without Diabetes. N Engl J Med. 2023;389(24):2221–2232. doi:10.1056/NEJMoa2307563. PubMed PMID: 37952131. Link{target="_blank" rel="noopener noreferrer"}
-
U.S. Food and Drug Administration. FDA Clarifies Policies for Compounders as National GLP-1 Supply Begins to Stabilize. February 2025. Link{target="_blank" rel="noopener noreferrer"}
-
Lasker Foundation. 2024 Albert Lasker Basic Medical Research Award: GLP-1 based drugs. 2024. PubMed PMID: 39352127. Link{target="_blank" rel="noopener noreferrer"}
Disclaimer. Statements in this article have not been evaluated by the Food and Drug Administration. This compound is not intended to diagnose, treat, cure, or prevent any disease. Sparta Labs sells research-use-only materials. Content is provided for educational and informational purposes only and does not constitute medical advice. Consult a qualified medical professional for any health concerns.
Frequently asked questions
What is semaglutide?
Semaglutide is a synthetic 31-amino-acid analogue of the human incretin hormone glucagon-like peptide-1 (GLP-1), engineered to resist enzymatic degradation and bind serum albumin. It was developed by a research team at Novo Nordisk and described in the peer-reviewed literature in 2015. It is classified as a GLP-1 receptor agonist.
Why does semaglutide have such a long half-life compared with native GLP-1?
Native GLP-1 is cleaved within about two minutes by the enzyme DPP-4. Semaglutide carries an alpha-aminoisobutyric acid substitution at position 8 that blocks that cleavage, plus a C18 fatty-diacid side chain that binds reversibly to circulating albumin. Lau and colleagues reported that these features together extend its terminal half-life in humans to roughly 165 hours.
How is semaglutide different from liraglutide?
Both are acylated GLP-1 analogues from the same Novo Nordisk research lineage, but they differ in their lipid anchor. Liraglutide uses a shorter C16 fatty-acid chain attached by a single glutamate spacer, while semaglutide uses a longer C18 diacid with a bifunctional gamma-glutamate and mini-PEG linker. Knudsen and Lau describe this change as the basis for semaglutide's markedly longer reported half-life.
What is the SNAC additive in oral semaglutide?
SNAC is sodium N-[8-(2-hydroxybenzoyl)amino]caprylate, an absorption enhancer co-formulated with semaglutide in its oral tablet form. Published research describes SNAC as raising local gastric pH and transiently increasing membrane permeability, which facilitates absorption of the peptide across the gastric epithelium. It made semaglutide the first orally administered GLP-1 receptor agonist to receive FDA authorization.
Is semaglutide FDA approved?
Yes, in specific pharmaceutical formulations. A subcutaneous formulation was authorized in December 2017 and an oral tablet in September 2019 for glycemic management in type 2 diabetes; a higher-concentration subcutaneous formulation followed in June 2021 for chronic weight management, with a cardiovascular indication added after the SELECT trial. Research-grade material sold for laboratory use is a distinct, non-pharmaceutical category.